
马宁宁教授:小型化抗体偶联药物及类似物的研究进展
作者:马宁宁课题组
日期:2024-06-16
浏览次数:

马宁宁,沈阳药科大学特聘教授,国家特聘专家,曾在美国辉瑞公司从事重组蛋白药物和单克隆抗体开发,2008 年获得辉瑞全球研发最高奖项。回国后曾任中国医学科学院细胞工程研发中心副主任,北京义翘神州副总经理,领导开发了我国首个重组凝血八因子药物。马宁宁教授于 2014 年被引进沈阳药科大学,以本溪市大型仪器公共技术服务生物检测平台为基础组建辽宁省生物制药动物细胞培养工程技术研究中心,承担辽宁省科技厅计划项目——哺乳动物细胞培养关键技术及创新药物研发。获得辽宁省教育厅创新团队成绩、辽宁省教育厅高等学校攀登学者(2017)、大挑战 2015·青年科学家( 科技部与比尔及梅琳达·盖茨基金会联合颁发)。马宁宁教授在重组蛋白药物生产方面与齐鲁制药深度合作,进行 GLP1-Fc 融合蛋白细胞株、小试工艺、质量研究技术开发;开发细胞基质流感病毒的生产工艺技术,并将该技术转让于艾美生物疫苗技术集团;在抗体及其突变体开发应用上取得很好的进展,完成专利转让;承接 2019 年国家“重大新药创制”重大科技专项子课题。
马宁宁教授团队在双抗药物开发、ADC 定点偶联技术上取得很好的突破,与美国默沙东和美国德州大学合作开发了具备广谱中和活性的抗 CMV 双抗。团队攻克了 one-click(单步)ADC 药物定点偶联技术以及双抗 ADC 定点偶联技术,具有以下优势:1)既可实现抗体恒定区,也可实现可变区的定点偶联;2)偶联工艺极其简单,可单步完成;3)全抗定点偶联、抗体片段定点偶联和双抗定点偶联均可实现(相关专利:WO2021/03193、PCT/CN2023/103976)。
【摘要】在癌症治疗中,实体瘤的治疗效果一直是医药界关注的热点。临床上用于治疗实体瘤的靶向药物——抗体和抗体偶联药物存在其相对分子质量较大,导致穿透性有限的问题,在实体瘤治疗中无法发挥预期疗效,且由于其在体内的循环时间长,易对肝和其他组织产生脱靶毒性,限制治疗窗。使用小型化的偶联物或类似药物,如抗体片段偶联药物、支架抗体偶联物或多肽偶联物,将有利于药物快速穿透肿瘤组织,使毒素在肿瘤组织内迅速聚集,且相比于传统抗体偶联药物,小型化偶联物通过肾脏代谢比率增加,从而降低药物因长时间体循环导致的不良反应。但是其过短的半衰期会造成进入肿瘤的实际药量减少,因此也衍生出不同半衰期延长方法,以调节药物半衰期,增加药物进入肿瘤组织的总量,提高治疗效果。通过对不同的小片段技术和代表性药物的临床前或临床进展情况进行介绍,以及对其发展方向进行讨论,以期为小型化抗体偶联药物及类似物的研发提供参考。
1.概述
肿瘤化疗源自于 20 世纪 40 年代,发展初期,药学研究者采用氮芥和叶酸拮抗剂治疗血液系统恶性肿瘤,在随后几年里发展出使用烷化药物和抗代谢物等化疗药物的疗法。癌症化疗虽然能延长肿瘤患者的缓解持续时间(duration of response, DOR),但仍面临药物非特异性毒性高、治疗窗过窄以及肿瘤耐药性增加等限制因素。20 世纪 70 年代,基于单克隆抗体(monocloning antibody,mAb)的疗法开始出现,其结构如图 1 A 所示, mAb 可以特异性结合癌细胞上的抗原,靶向肿瘤细胞,减少非特异性毒性,通过改变信号传导模式或引起免疫反应发挥疗效。同时,在接下来十几年中也有研究尝试将抗体与细胞毒性药物、放射性药物或免疫毒素等进行偶联,构建抗体偶联药物(antibody drug conjugate,ADC),通过抗体部分实现对肿瘤组织的精准靶向,将毒素递送至肿瘤组织,减少全身暴露和毒性,提高疗效[1]。目前,有 100 多种 ADC 正在进行临床试验,但只有 14 种 ADC 被美国食品药品监督管理局(FDA)批准用于临床。在 14 种批准的药物中,7 种用于治疗血液系统恶性肿瘤,7 种用于治疗实体瘤。由于靶向毒性、脱靶毒性和疗效不足等情况,大多数 ADC 在临床阶段被终止[2]。
一直以来,药物在实体瘤中的穿透性都是影响药效的重要因素。抗体和 ADC 在给药后,药物在肿瘤中的摄取剂量峰值不足给药剂量的 0.1%[3],未达到该类药物的预期效果。一项关于表皮生长因子受 体(epidermal growth factor receptor,EGFR)抗体 ch806 的Ⅰ期临床试验研究表明,患者在药物注射 5 ~ 7 d 后,肿瘤摄取才达到峰值[4],存在肿瘤摄取慢的问题。其原因可能是抗体等大分子药物穿透实体瘤,需要克服血管屏障、细胞外基质屏障和抗原细胞屏障这三重障碍,即病理情况下实体瘤内血流不畅,造成有效灌注量过少;间质中细胞外基质过于致密,导致血浆对流转运速率降低;同时,靶点在肿瘤组织中的表达异常高,需要大剂量 ADC 去饱和肿瘤边缘形成的“结合部位屏障”(binding site barrier,BSB)[5],采用相对分子质量较小的骨架偶联毒素药物是解决该问题的一种出路,已有模型比较 mAb(相对分子质量 150 000)、抗原结合片段(fragment of antigen binding,Fab)(相对分子质量 50 000,其骨架如图 1C 所示)、F(ab)2(相对分子质量 100 000)以及单链抗体(single chain variable fragment,scFv)(相对分子质量 27 000,其骨架如图 1D 所示)在不同组织中的穿透性,结果显示穿透性与其分子大小呈负相关,但模型未比较他们在实体瘤中的穿透情况[6],也有研究采用异种移植小鼠肿瘤模型考察分子量对于渗透性的影响,比较 scFv、sc(Fv)2、mAb 的渗透性,观察到 scFv 摄取和渗透最快,mAb 最慢[7]。以双环肽(Bicycle)(见图 1 I)为骨架的多肽偶联药物(peptide drug conjugates,PDC)BT5528 在临床前研究中显示出比免疫球蛋白 G(immunoglobulin G,IgG)型抗体 MEDI-547 更快的肿瘤蓄积,其达峰时间仅为 8 h,远低于 IgG 型 ADC 近 24 h 的达峰时间;且 BT5528 肿瘤与血浆药物浓度比可达 100 : 1 以上,而 IgG 型 ADC 该比值始终低于 1 : 1;同时在异种移植小鼠肿瘤模型中显示,当肿瘤体积大于 1 000 mm3 时, PDC 能实现完全抑制,而 IgG 型 ADC 仅能实现部分抑制[8]。根据临床前结果显示,降低靶向偶联物的相对分子质量,可以增加其实体瘤的穿透能力,提高肿瘤组织与血浆组织药物浓度比值,降低不良反应的程度。
除了穿透性不同,IgG 型 ADC 与相对分子质量更小的偶联物在药物代谢动力学(pharmacokinetic, PK)上也有明显的差异。IgG 型抗体由于新生儿 Fc 受体(the neonatal Fc receptor,FcRn)的转运和保护机制,血浆半衰期得到延长[9],而片段化抗体,由于无 Fc 结构,半衰期主要取决于流体动力学直径(hydrodynamic diameter,HD),HD 小于 5 ~ 6 nm 的蛋白会随尿液快速代谢;相对分子质量在 30 000 左右的 scFv,HD 为 5.3 nm,有 74%经肾代谢,血浆半衰期为 11 min,全身半衰期为 1.4 h;相对分子质量为 60 000 的 Fab,HD 为 6.0,仅有 9%经肾代谢,血浆半衰期为 28 min,全身半衰期为 1.4 h;相对分子质量为 60 000 的 sc(Fv)2 和相对分子质量为 120 000 的(sc(Fv)2)2 HD 均大于 6 nm,高于肾脏排泄阈值,血浆半衰期分别为 78 和 170 min,全身半衰期分别为 5.1 和 8.9 h,均低于 IgG 型抗体的 330 min 和 730 h[10],PDC 类药物主要利用肾进行代谢,在人体内的半衰期不超过 4 h。IgG 型 ADC 的较长半衰期,确保了更高的生物利用度,但也引起了更多负面影响,其 PK 曲线类似平台期,可以在非靶器官中引起毒素释放和毒性。ADC 通常通过肝代谢消除,导致在肝和胃肠道中也会存在有效载荷的释放,引起剂量限制性毒性[11]。而以 PDC 为代表的低分子量药物,利用肾代谢,其血浆半衰期短,可能会引起生物利用度过低,但系统暴露度低、不良反应小。PDC 的较短的血浆半衰期与该类分子的高肿瘤穿透性相结合,能达到高肿瘤蓄积比,即高肿瘤内药物浓度和低血浆药物浓度。PDC 药物 BT5528,在临床前和临床研究中证明了其相比于同靶点 IgG 型 ADC 药物 MEDI-527,在治疗指数上明显提高[12]。除了 ADC 和 PDC 2 种极端代谢谱,小分子量偶联物可以通过白蛋白结合域融合等技术调节半衰期,在实现高肿瘤蓄积比,拓宽 ADC 药物的治疗窗的同时,提高生物利用度。
对于作用于实体瘤的药物来说,穿透性和 PK都是需要考虑的因素,其变化都会对药物的治疗窗产生影响,本文总结了目前报道的用于治疗实体瘤的各类小片段偶联物的靶向部分的设计、毒素、偶联技术、半衰期延长手段,以及该类药物现有的临床前药效学和药代动力学结果,以期为小型化抗体偶联药物及类似物的研发提供参考。

2.小片段偶联技术
2.1 抗体片段偶联物
抗体片段偶联物(antibody fragment drug conjugate,AFDC),是 scFv、Nb、Fab、diabody 等通过基因工程方式改造抗体的抗体片段与毒性药物偶联构建的药物。从偶联工艺方面,AFDC 的偶联技术主要集中在以下几类:1)通过连接子上的苄基鸟嘌呤(O6-benzylguanine,BG)与末端 SNAP 标签(O6-甲基鸟嘌呤-DNA 甲基转移酶改造的蛋白)偶联;2)通过分选酶 A(sortase A),将带 NH2- GGG 的连接子与末端带 LPETG-COOH 的 AFDC 偶联,形成 LPETGGG 的结构;3)使用二硫键桥连的连接技术进行偶联,即还原抗体中的二硫键后,采用与半胱氨酸选择性反应的试剂如二硫代马来酰亚胺(dithiomaleimide,DTM)对 AFDC 的二硫键进行重新桥连的同时引入细胞毒性药物;4)利用 AFDC 末端的半胱氨酸与马来酰亚胺偶联。除此之外,也有利用非天然氨基酸引入叠氮进行偶联和使用带[ 乙二胺铂(Ⅱ)] 2+连接子与末端二硫键反应的相关报道。除抗体片段上赖氨酸与胺基进行随机偶联的非定点偶联技术外,AFDC 的定点偶联部位大多集中在抗体 C 末端,目前 AFDC 涵盖有放疗药物、光疗法药物和小分子毒素偶联物,多种连接子均有报道,部分药物汇总可见表 1。
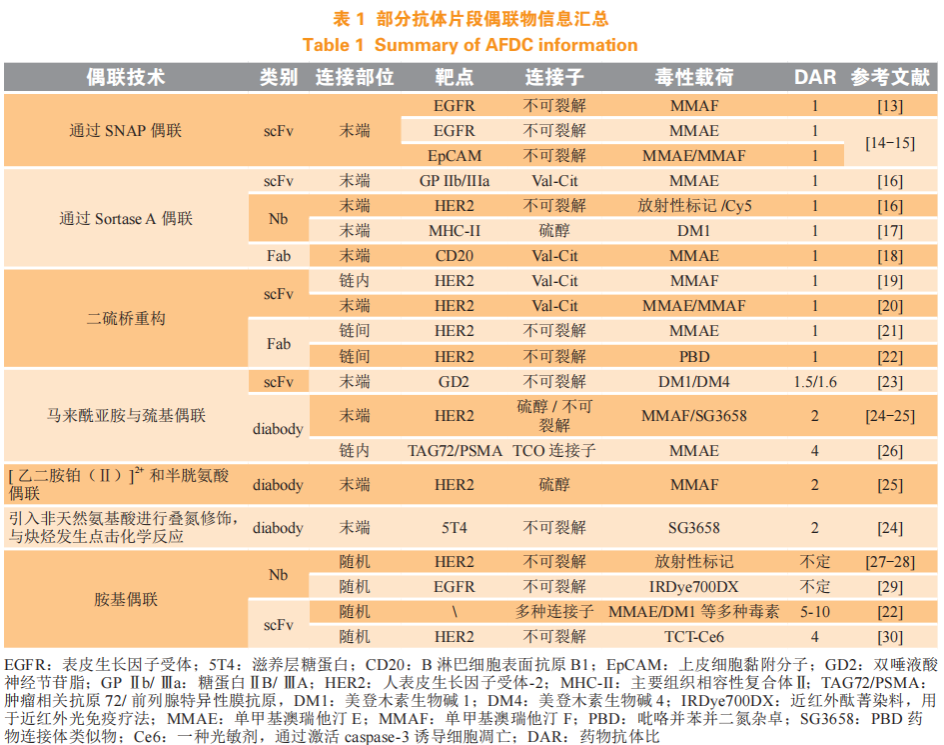
关于体内抗肿瘤活性方面,比较 AFDC 与 IgG 型 ADC 抗肿瘤活性的研究较少,目前已报道 Fab 与 IgG-ADC 的比较中,Fab 并没有显示出更强的抗肿瘤活性,DAR 值为 2.8 的曲妥珠全长抗体偶联物与 DAR 值为 1 的 Fab 偶联物进行 BT474 小鼠肿瘤模型的药效分析时发现,Fab 肿瘤抑制效果不如前者,并且在治疗后期出现肿瘤复发[21],在 CD20 相关抗体的小鼠肿瘤模型中也发生了类似的现象[18],这可能与 Fab 在血浆中代谢较快相关。
关于 AFDC 的半衰期延长(half-life extension, HLE)策略,常见为以下几种方式——融合人血清白蛋白(human serum albumin,HSA)、融合白蛋白结合结构域(albumin binding domian,ABD)、对抗体进行聚乙二醇化,三者利用的原理有所区别。融合 HSA 的结构可见图 1J,延长半衰期的基本原理为,HSA 结构域Ⅲ与 FcRn 相互作用,从而延长半衰期[31],也有报道称 HSA 不会被肾小球滤过 [10];ABD 融合结构可见图 1K,半衰期延长基本原理为融合 ABD 的抗体与人血清白蛋白发生动态结合,通过白蛋白参与 FcRn 介导的再循环,从而延长体内半衰期[32];而抗体的聚乙二醇化主要通过改变电荷或增加分子大小,来降低肾清除率,从而改善 PK[33]。Li 等[24]考察了 diabody 与 ABD 融合和聚乙二醇化 2 种 HLE 方式,两种 HLE 均能将抗体偶联物的半衰期从几分钟延长到几天,其在乳腺癌异种移植模型的体内研究结果表明,ABD 融合偶联物比聚乙二醇化偶联物具有更高的体内肿瘤生长抑制活性和更好的耐受性,前者在 19 nmol · kg-1 剂量下,显示出 96%的肿瘤抑制率。Xenaki 等[25]考查了单剂量给药下 ABD 融合的 AFDC 11A4-ABD-malAF、11A4-ABD-Lx-AF 和非融合形式的 11A4-malAF、11A4-Lx-AF 的体内活性,前两者半衰期较后者提高了 14.8 倍,出现明显肿瘤抑制,而未融合组肿瘤持续增大,且生存期明显短于前两者,说明 HLE 可以在一定程度上增强小鼠肿瘤模型的药效。除了上述 HLE 方式,使用赖氨酸偶联方法构建高 DAR 值的 AFDC 也可能引起肾代谢变化,半衰期最高可增加 10 倍,但其体内药效没有报道[22]。目前上述 HLE 改造的偶联物均处于临床前阶段,临床表现有待进一步研究。
2.2 非抗体支架蛋白药物偶联物
2.2.1 亲和体偶联物 亲和体是一类由 58 个氨基酸构成的、具有反平行的三螺旋折叠的小分子量蛋白,是一种非免疫球蛋白形式的亲和蛋白,其相对分子质量为 6 500 左右[34],结果如图 1 H 所示。目前,采用第一代 Affibody 支架构建设计的靶向人表皮生长因子受体-2(human epidermal growth factor receptor 2,HER2)的正电子发射型计算机断层显像(positron emission computed tomography,PET)诊 断试剂 ABY-025 已进入Ⅱ期临床阶段[35]。由于相对分子质量较小,这类药物可以实现在肿瘤中快速分布和在肾脏中的快速代谢,是一类理想的靶向诊断试剂,但作为治疗性药物来说,存在着肾小管重吸收的问题[36],如果制备成偶联物,可能会增加其肾脏不良反应。在构建 Affibody 为骨架的靶向放疗药物时,Affibody AB 公司的研发团队将 Affibody 骨架与 ABD 融合,从而延长血浆半衰期并减少肾脏的摄取[37],同时将其中的 ZHER2:324 优化为第二代支架 ZHER2:2891,可增加其亲水性和热稳定性[34],且比较了 2 种在 ZHER2:2891 C 端融合 ABD 的药物——放射性药物[ 177Lu]Lu 偶联在 C 末端的 ABY-027 和偶联在 ABD 第 14 位的 ABY-271,筛选出肿瘤肾脏分布比更高的[ 177Lu]Lu -ABY-027[38],目前正在进行与曲妥珠联合用药的体内活性研究[39],除此之 外,根 据 Affibody AB 官网显示[35],[ 177Lu]Lu -ABY-271 正在临床申报阶段[35]。瑞典乌普萨拉大学 Orlova 团队也利用该类骨架,开发 Affibody 偶联物,在骨架末端引入半胱氨酸,制备美登木素生物碱 1(mertansine DM1,DM1)、一甲基澳瑞他汀 E (monomethyl auristatin E,MMAE)和一甲基澳瑞他汀 F(monomethyl auristatin F,MMAF)偶联药物[40],与 ABD 进行融合,将部分肾脏代谢转变为肝代谢[40];在骨架末端半胱氨酸前,使用 3 个谷氨酸作为间隔以降低动物体内肝摄取[41-42],构建 DAR 为 1 和 3 的 Affibody 偶联物,目前 DAR 为 3 的药物虽体内药效较好,但肝、脾、骨的非特异性摄取更多;也考察了不同结构域排列[43]、骨架与 ABD 间不同蛋白连接肽连接子长度的影响[44],目前,该偶联物还在进行临床前优化阶段,没有明确的最优分子。
2.2.2 预设计锚蛋白重复蛋白偶联物 DARPins 是一类小的单结构域蛋白(相对分子质量为 14 000 ~ 21 000),其利用天然存在的锚定重复蛋白设计而成[45],其结构如图 1E 所示。DARPins 可借助大型文库筛选,从而得到高稳定性、高可溶性和高产量的蛋白序列,其结构不需要翻译后修饰,可直接采用大肠杆菌表达[45]。根据 Molecular Partners 官网显示,其产量能达到 15 g · L-1 ,稳定性能支持其在 4℃环境放置数年[46]。目前,该公司研发的以血管内皮生长因子(vascular endothelial growth factor, VEGF)为靶点的非偶联 DARPin 药物 Abicipar,由于给药后观察到的眼内炎症率相比于其他同靶点药物更高,其上市申请在 2020 年被美国 FDA 驳回[47],同时,新型冠状病毒感染期间 FDA 对 ensovibep 的紧 急使 用授 权(Emergency Use Authorization, EUA)申请已于 2023 年 1 月 25 日被撤回,退回到临床开发阶段,除此之外,MP0317、MP0533 2 个药物正在进行Ⅰ期临床试验[48]。关于 DARPin 偶联的放疗药物,Molecular Partners 公司在 2021 年已与诺华合作,团队于 2023 年欧洲核医学协会(European Association of Nuclear Medicine,EANM)年会和美国癌症研究协会(American Association for Cancer Research,AACR)年会上公布了其研究结果,其在 DARPins 骨架部分进行优化,可显著降低肾小管重吸收,将肾代谢药时曲线下面积(area under curve, AUC)降低 76%,同时对肿瘤内药物浓度无明显影响[49], Molecular Partners 也公布了 3 种 HLE 改造 DARPins 的结果,但目前没有公布其 HLE 方式,其中,与未改造蛋白相比,最佳分布的 DARPin HLE 4 h 肿瘤摄取量由 3.4% ID· g-1 升高至 8.5% ID· g-1 ,血浆含量由 0.02%ID·g-1 升高至 22.9 % ID·g-1 ;24 h肿瘤摄取量由 2.9% ID · g-1 升高至 19.1% ID · g-1 ,血浆含量由0.01% ID·g-1 升高至22.9 % ID·g-1[50]。此外,关于 DARPins 偶联物,2019 年 ImmunoGen 公司在 2019 年的 AACR 会议上公布了抗 EGFR DARPins PSC009、PSC106 特异性偶联 DGN549 的结果,显示其在 5 mg · kg-1 的剂量下,对异种移植模型的肿瘤抑制率能达到 100%,且体重减少小于 20%[51],目前,该类药物偶联物均处于临床前研究阶段,无临床进展。
2.2.3 Centyrin 偶联物 Centyrin 是一种相对分子质量为 10 000 的无硫醇支架,基于Ⅲ型纤连蛋白结构域设计,具有高亲和力、高热稳定性和化学稳定性[52],其结构如图 1G 所示。目前,该支架报道的偶联物仅有抗 EGFR 支架偶联物,Goldberg 等[53]向 Centyrin 支架中引入半胱氨酸,实现与 MMAF 的偶联,目前,该支架偶联物主要用于递送寡核苷酸药物,无体内实验结果的相关报道。
2.2.4 Abdurin 偶联物 Abdurins 是一种基于工程化 IgG CH2 结构域的小型抗体样支架,具有与 FcRn 受体结合的能力,有较长的循环半衰期,相对分子质量仅为 15 000。利用该平台筛选的抗肾上腺素 A 型受体 2(tyrosine protein kinase receptor A2, EphA2)支架在注射 4 h 后即可定位于异种移植模型肿瘤中,并在肿瘤中持续积累长达 48 h 以上[54]。但该支架偶联后存在一定问题,2019 年蛋白工程峰会上,Peretti 公布了载荷为 vcMMAE 的 Abdurin 偶联物,显示 DAR 为 1 的抗体在 PC3 肿瘤异种移植模型中的抑制能力较差,将 DAR 提高到 2 后,偶联物与靶标以及FcRn的亲和力出现明显的下降[55],此后无该非抗体支架偶联物的进展公布。
2.3 多肽偶联物
根据 FDA 定义,多肽为 40 个或更少的氨基酸组成的、相对分子质量为 500 ~ 5 000 的聚合物。多肽偶联物(peptide drug conjugates,PDC)为多肽与细胞毒性药物偶联产生的一种治疗性药物或诊断试剂,根据其作用机制可分为细胞穿透肽(cellpenetrating peptides,CPP)和 细胞 靶向 肽(celltargeting peptides,CTP),也有少部分的 PDC(如欧洲上市的 Melflufen)同时具有 CPP 和 CTP 的特性[55]。Cybrexa Theraputics 公司的 alphalexTM 技术为 CPP 的一个成功代表,目前该平台下的 SN-38 偶联物候选分子 CBX-12 已进入临床阶段[56],该技术利用肽在低 pH 环境下和正常 pH 环境下结构不同引起的细胞穿透能力的差异,即肽在低pH下形成α-螺旋,可以穿透细胞膜,将毒素如 MMAE、DM1、拓扑异构酶抑制剂等带入到细胞内,正常 pH 环境下不具有穿透能力[57];Melflufen 这类药物发挥作用的主要机制为利用亲脂性的药物穿透细胞膜,进入肿瘤细胞后,分子上氨肽酶结合域能作为底物,和肿瘤细胞特异性表达的氨肽酶和酯酶反应,释放亲水烷基化剂,引起细胞毒性[58],这 2 类药物虽具有一定的靶向性,但作用机制和本文讨论的与 ADC 作用机制类似的小片段偶联物作用机制差别较大,故本文不再进行讨论,仅对 CTP 进行讨论。
从归巢肽部分的来源来看,可将 CTP 分为 2 类,一类为使用天然配体多肽或天然配体多肽改造的稳定类似物构建的传统 PDC,目前进入临床阶段的 PDC 大部分都属于该类;另一类为通过噬菌体库等手段,筛选出的非天然多肽与毒素偶联构建的 PDC。
使用天然配体多肽或将其改造为稳定类似物构建的 PDC 靶点主要集中在整合素(integrins)、生长抑素受体(somatostatin receptors ,SSTRs)、促性腺激素释放受体(gonadotropin-releasing hormone receptor,GnRH-R)、铃蟾肽受体[bombesin(Bn) receptor,Bn receptor][59],也有少部分低密度脂蛋白受体相关蛋白-1(lipoprotein receptor-related protein 1,LRP-1)[60],分拣蛋白 1(sortilin-receptor 1, SORT1)相关药物正在研究[61],这些靶点对应的多肽和药物如表 2 所示。该类 PDC 有部分存在亲和谱较大的情况,如目前常用来靶向整合素αvβ3 的由精氨酸-甘氨酸-天冬氨酸组成的序列多肽(RGD)基序,在纤维蛋白原、纤维连接蛋白、凝血酶原、替纳辛和其他糖蛋白中都有存在,可以和超过 24 种整合素结合[59];目前已进入临床的靶向 SSTR2 的 PDC PEN-221,其骨架奥曲肽为 SST 类似物,对 SSTR2 的亲和力高达 16 pmol·L-1 ,但其同样对同家族的 SSTR1、SSTR3、SSTR5 也具有纳摩尔级别的亲和力(310、19.8 和 9.6 nmol · L-1 )[62],这可能是目前该类多肽选择与低毒性药物(如紫杉醇、阿霉素)或放疗药物(如 177Lu、18F、68Ga、90Y、99mTc、 111In)偶联的原因。
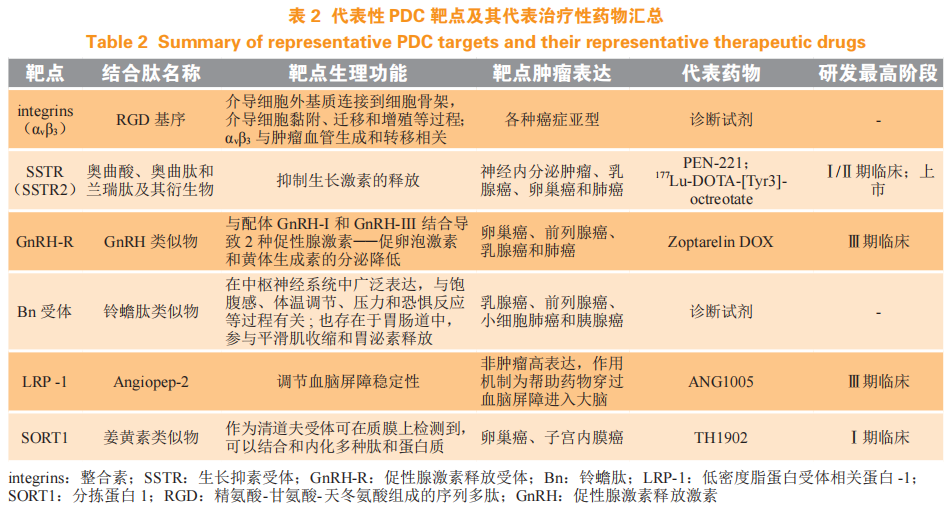
使用菌体库、肽库筛选抗体,计算机辅助药物设计的方法发现抗体,或将上述方式相结合,可能得到选择性更好的多肽,同时也能实现对不同靶点的定向筛选,使 PDC 不止局限在上述靶点中,如 Bicycle Theraputics 公司采用噬菌体库筛选的偶联 DM1 的 BT1718 和偶联 MMAE 的 BT5528、BT8009,对人、大鼠、小鼠和灵长类动物的目标蛋白均有纳摩尔级亲和力,对同家族其他蛋白无亲和力,降低了脱靶毒性,且在临床前研究中证明,与 IgG 型 ADC 不同 ,上述 3 种药物在 1 000 mm3 肿瘤大小的小鼠肿瘤模型中均有良好的抗肿瘤活性[63-66],目前,这 3 种药物均进入Ⅰ/Ⅱ期临床阶段[12]。同样也有利用文库或从头设计方法得到的多肽处于临床前研究阶段, Wang 等[67]使用商业化 OBOC 肽库筛选出 LLC2B 多肽,通过二硫化物或马来酰亚胺连接子与 DM1 偶联,构建 LLC2B-SS-DM1 和 LLC2B-Mal-DM1,在小鼠肿瘤模型中,DM1 当量为 2.0 和 4.0 mg · kg-1 的 LLC2B-Mal-DM1 和当量为 0.5 mg · kg-1 的 LLC2B-SS-DM1 都具有明显的抗肿瘤作用,在 2 种 PDC 中,LLC2B-SS-DM1 比 LLC2B-Mal-DM1 具有 更好的抗肿瘤效果,但没有采用 1 000 mm3 肿瘤对其进行进一步验证;Geng 等[68]采用计算机辅助药物设计和肽库相结合进行定向筛选的方式,设计出 16 聚肽库基本骨架,将筛选库库容缩小到 72 个肽序列,后利用该肽库筛选出亲和力为 18.6 nmol· L-1 的抗 HER2 多肽, 并证明其在 BT474 体内外模型中的高穿透性;Zhou 等[69]直接利用 HER2 与曲妥珠的共结晶结构从头设计多肽偶联物,筛选出亲和力为 2.555 μmol·L-1 的多肽,将其偶联喜树碱抑制剂,设计出有杀伤毒性的 PDC。
3.结语与展望
治疗窗窄是制约 ADC 药物发展的一个主要因素,而实体瘤的低通透性是造成 ADC 药物治疗窗窄的一个重要原因。实体瘤的低通透性一方面造成仅有极少量药物达到作用部位,限制了药效;另一方面,为促进药物进入肿瘤组织,ADC 需要长期保持高血药浓度,增加了正常组织的暴露度,对 ADC 药物形成剂量限制。降低传统 ADC 药物的分子量和水动力半径,是提高实体瘤通透性的一个有效手段。
目前,在小型化 ADC 及其类似物中只有靶向放疗药物上市,但临床和临床前药物开发呈现出百花齐放的趋势,关于 AFDC 的研发出现了针对不同靶点、采用不同偶联工艺、选择不同 HLE 方式的偶联物;以 Affibody 和 DARPins 为首的非抗体支架偶联物目前也处于积极研发中,经 ABD 融合的 Affibody 相关放疗药物已进入临床申报阶段,同时,DARPins 偶联物在体外稳定性和体内分布上也展示出其肿瘤快速摄取和快速代谢的能力,且不同 HLE 展现出不同临床前效果;关于 PDC,以 Bicycle 为首的小片段偶联药物在临床前多个维度和部分临床结果上展露出靶向实体瘤的独特优势,相信不久的将来,能出现更多新的蛋白设计、偶联方式以及使用不同半衰期调节技术的小片段偶联药物,造福更多的患者。
辽宁省沈阳市沈河区文化路103号 | 邮编:110016
©2016 沈阳药科大学无涯创新学院 版权所有

 科研动态
科研动态

